
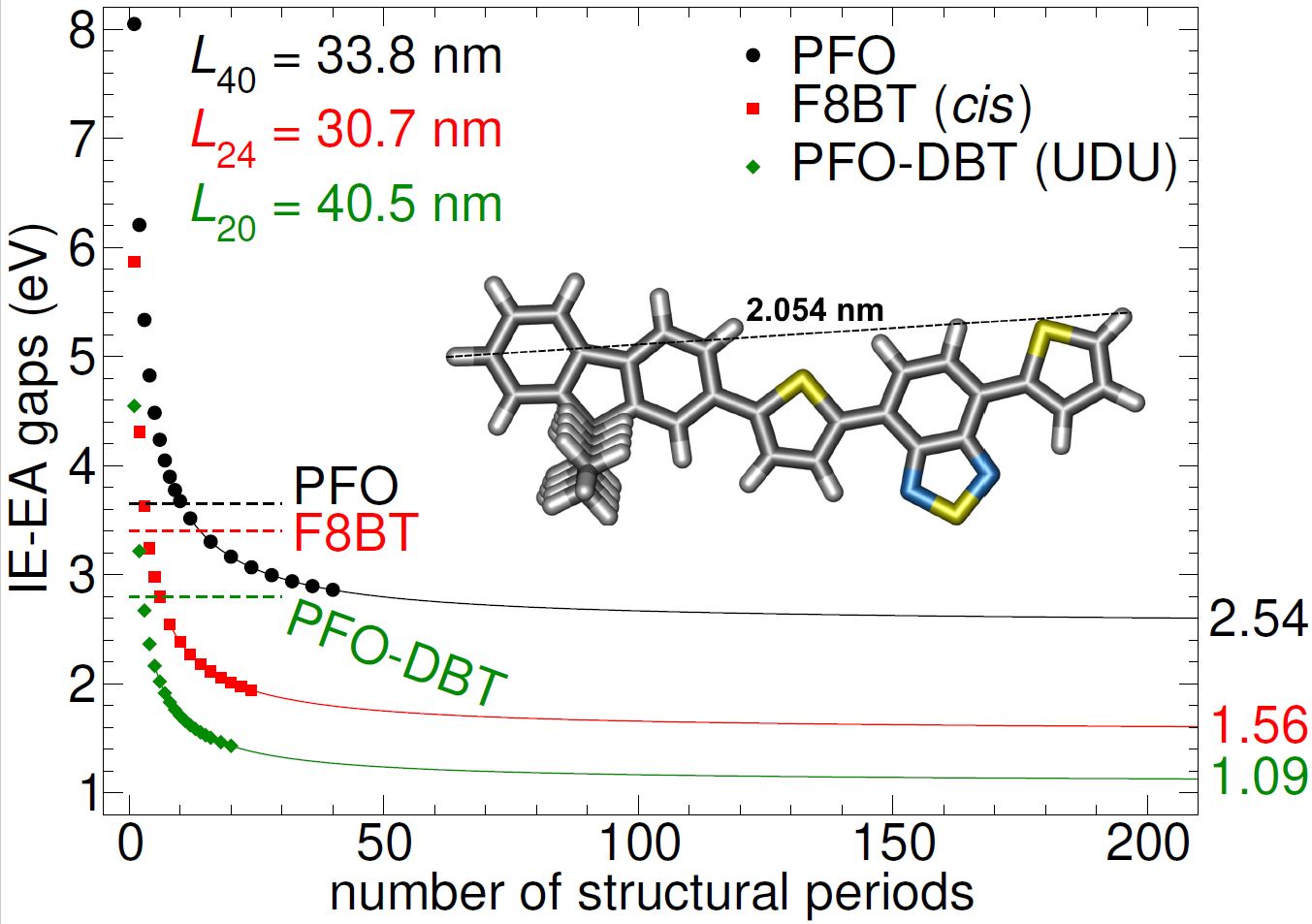
Devices for organic electronics and photonics, such as LEDs or solar cells, contain molecules whose key feature is an energy band gap (BG), similar to that in traditional semiconductors (e.g. silicon). For practical applications, the width of the BG is particularly important, as well as whether there are any energy levels within it, which may appear, for example, as a result of defects. These characteristics of the BG are closely related to the optical and electronic properties of materials. In addition to experimental methods, atomistic computer simulations are useful tools for studying these properties. This is also one of the research areas pursued at our institute. The output of such calculations often includes information about the atomic structure, as illustrated in Fig. 1, which specifically shows the PFO-DBT comonomer. We apply these computational approaches in cooperation with the Institute of Physics of the Slovak Academy of Sciences in Bratislava, where a unique method for measuring the density of electronic states of organic materials with a band gap has been developed. Our research using computer modelling helps to better understand the measured results, particularly through comparisons of calculated data with experimental results [1].
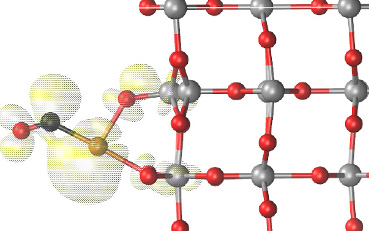
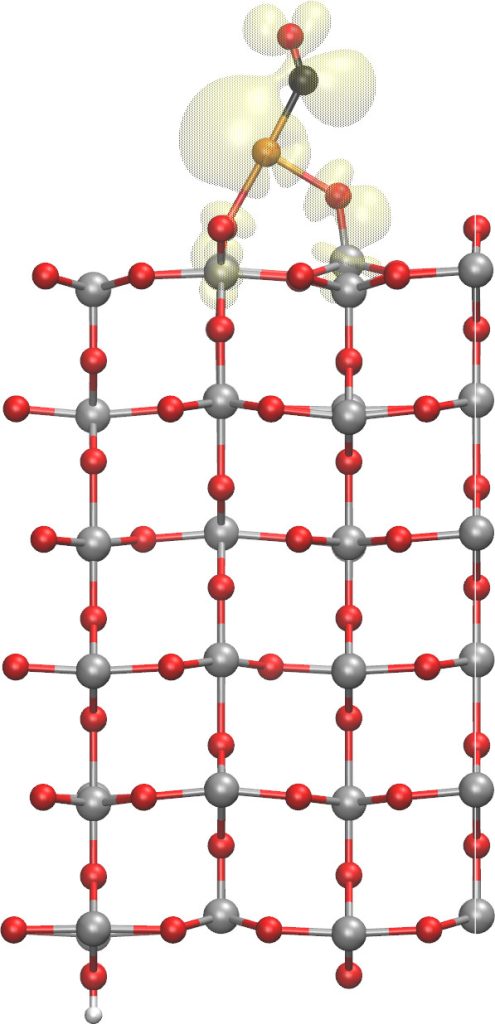
The second line of research being developed is cooperation with the Slovak Academy of Sciences and with international colleagues in Japan, China, and the United Kingdom. Within this collaboration, we investigate unique surface properties of rutile (TiO2) and its interactions. Our international partners carried out research using state-of-the-art KPFM instruments, which combine the capabilities of several microscopy techniques: atomic force microscopy (AFM), scanning tunnelling microscopy (STM), and surface electrostatic potential mapping. These instruments enabled, for example, manipulation of charge states of individual oxygen or gold atoms on the rutile surface (Fig. 2). The group at the Slovak Academy of Sciences and our institute both provide support for experimental results through calculations, simulations, and interpretation of measured data [2].
References
[1] Katarína Gmucová, Martin Konôpka, Karol Vegso, Peter Bokes, Vojtech Nádaždy, Tomáš Váry: Correlation between Molecular Stereostructure, Film Microstructure, and Electronic Structure of Polyfluorene and Fluorene-Based Alternating Copolymers F8BT and PFO−DBT, J. Phys. Chem. C 125, 8045 (2021),
https://doi.org/10.1021/acs.jpcc.0c10725
[2] Yuuki Adachi, Ján Brndiar, Martin Konôpka, Robert Turanský, Qiang Zhu, Huan Fei Wen, Yasuhiro Sugawara, Lev Kantorovich, Ivan Štich, Yan Jun Li: Tip-activated single-atom catalysis: CO oxidation on Au adatom on oxidized rutile TiO2 surface, Science Advances 9, eadi4799 (2023),
https://doi.org/10.1126/sciadv.adi4799